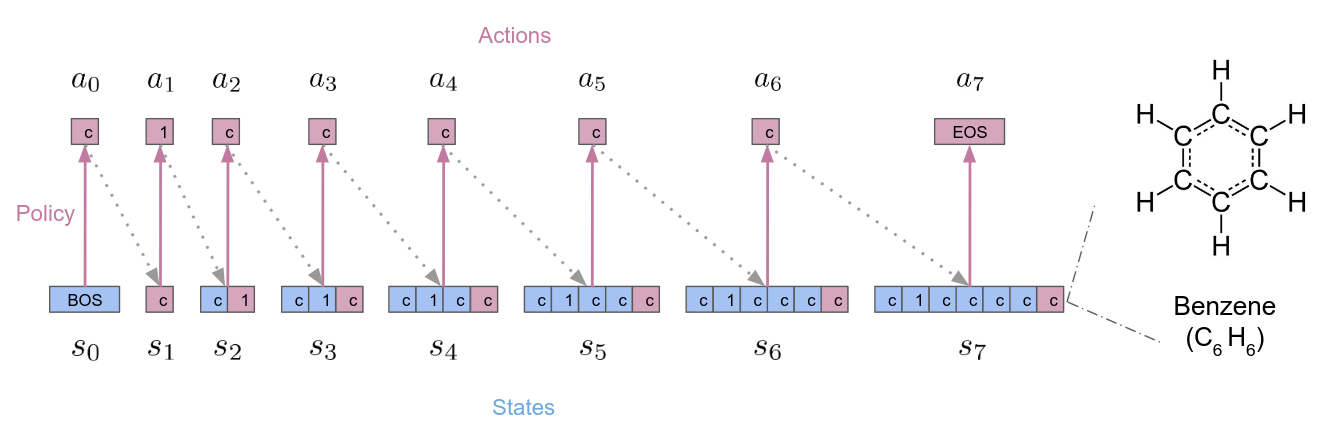
Reinforcement learning (RL) over text representations can be effective for finding high-value policies that can search over graphs. However, RL requires careful structuring of the search space and algorithm design to be effective in this challenge. Through extensive experiments, we explore how different design choices for text grammar and algorithmic choices for training can affect an RL policy's ability to generate molecules with desired properties. We arrive at a new RL-based molecular design algorithm (ChemRLFormer) and perform a thorough analysis using 25 molecule design tasks, including computationally complex protein docking simulations. From this analysis, we discover unique insights in this problem space and show that ChemRLFormer achieves state-of-the-art performance while being more straightforward than prior work by demystifying which design choices are actually helpful for text-based molecule design.
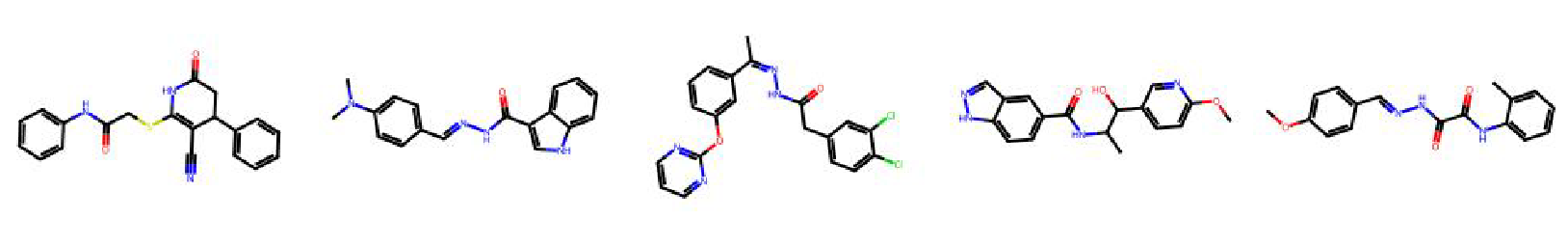
Molecules generated by ChemRLFormer by reward hacking the docking functions.
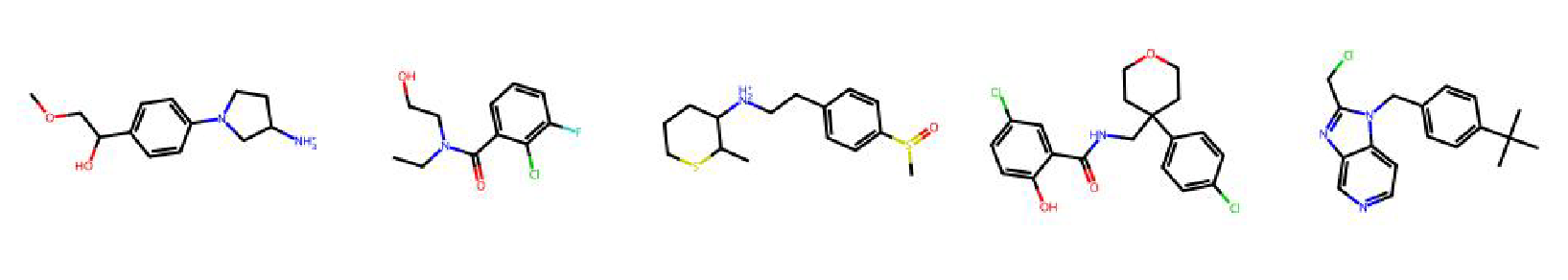
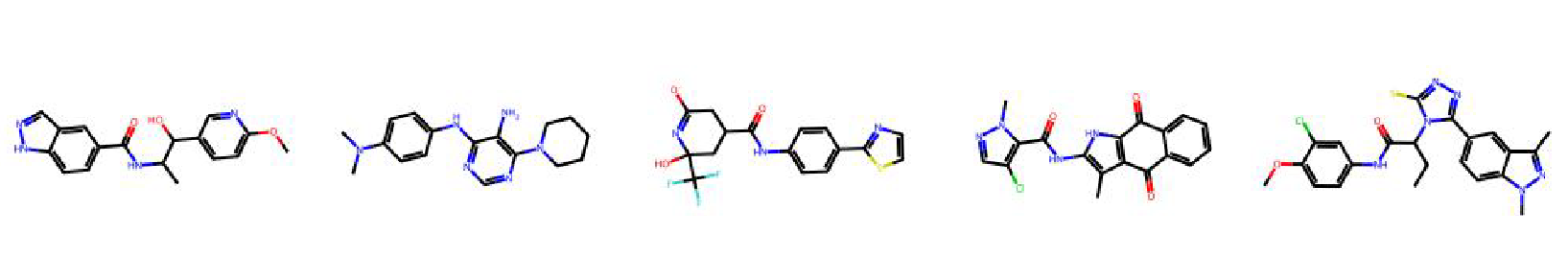
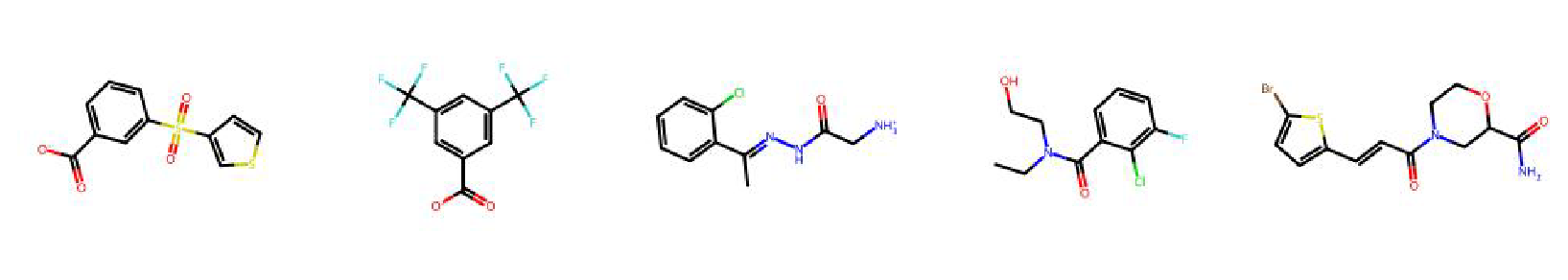
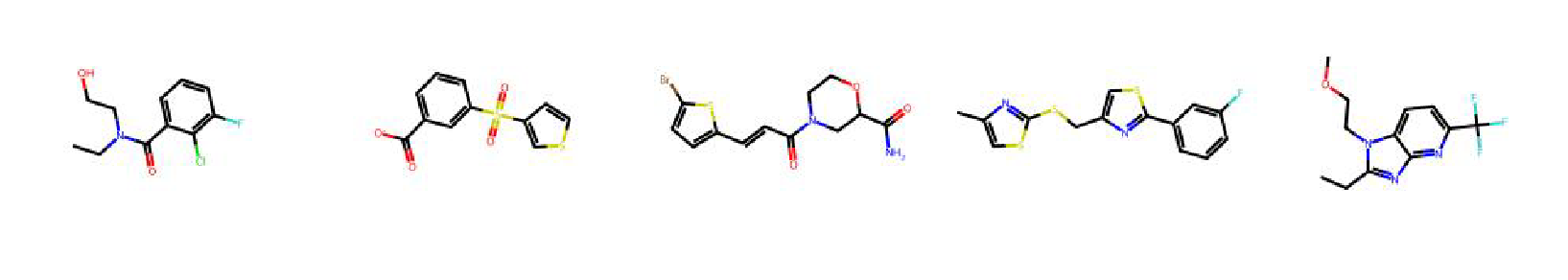
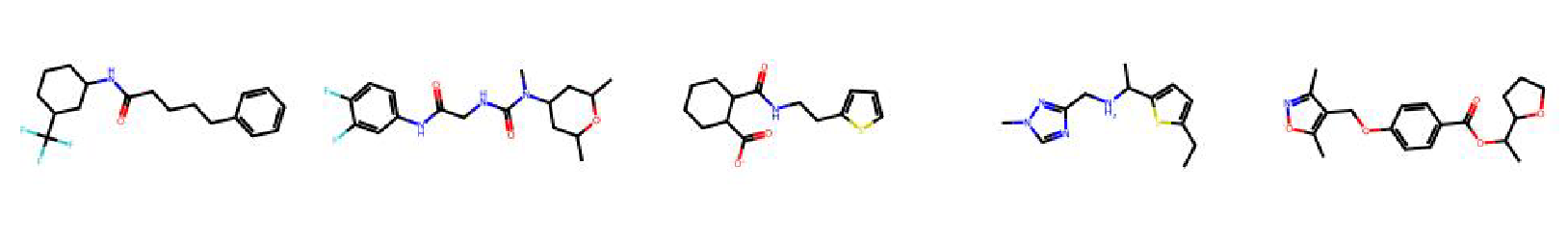
High value molecules generated by ChemRLFormer for various reward functions.
albuterol similarity
amlodipine mpo
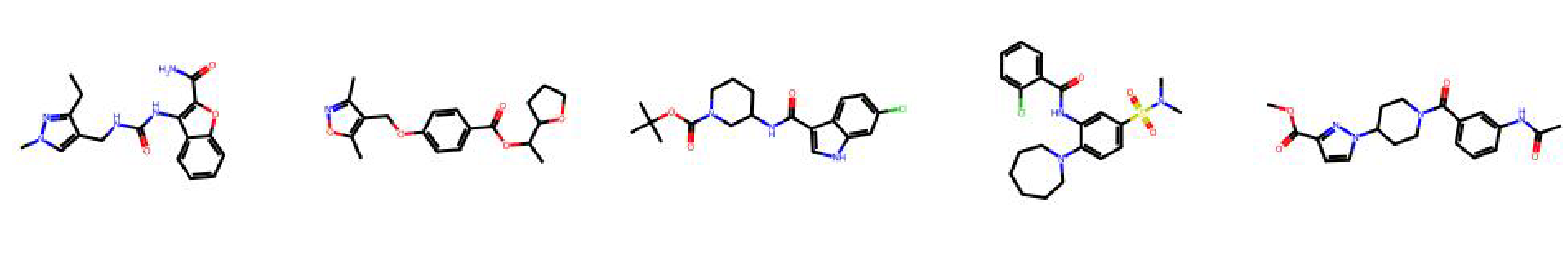
celecoxib rediscovery
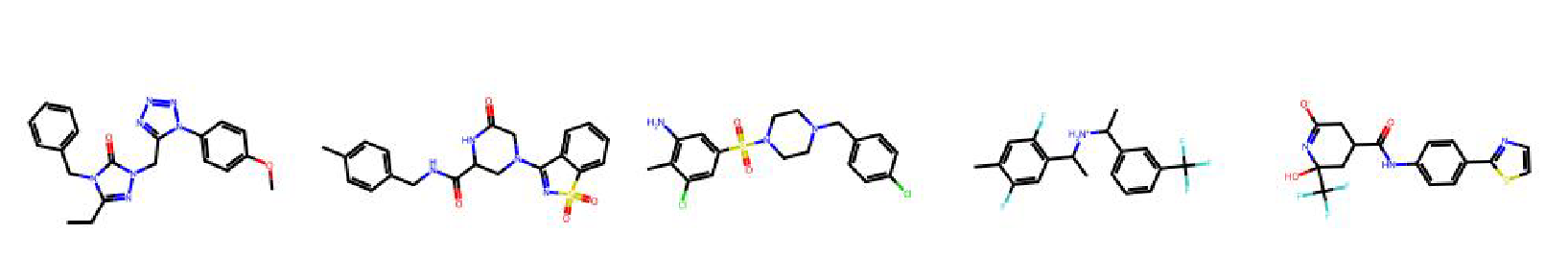
deco hop
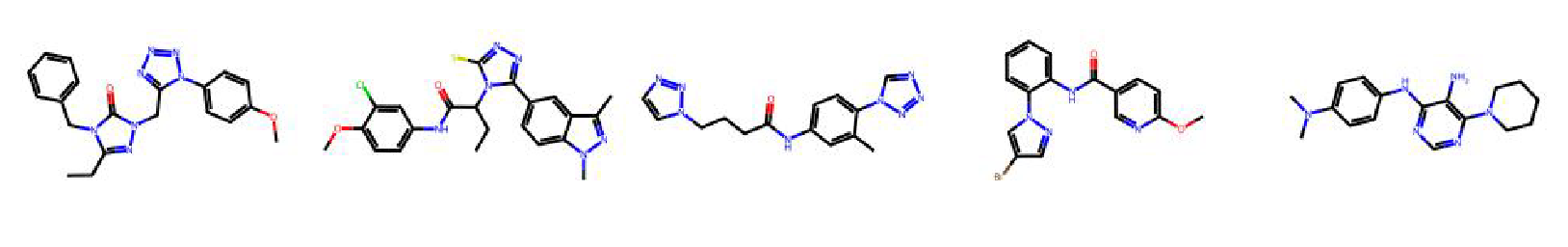
drd2
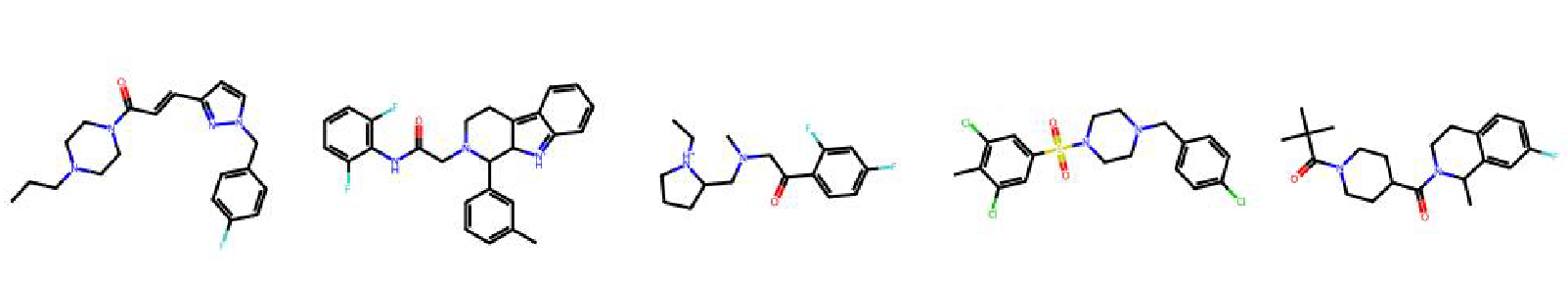
fexofenadine mpo
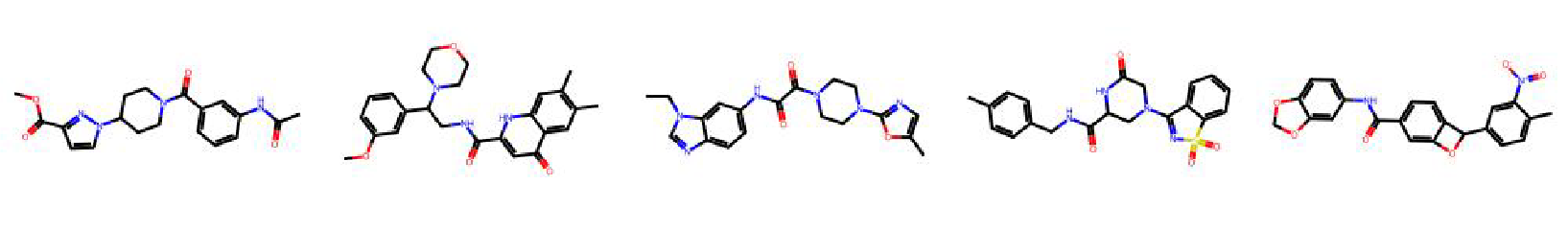
gsk3b
isomers c7h8n2o2
isomers c9h10n2o2pf2cl
jnk3
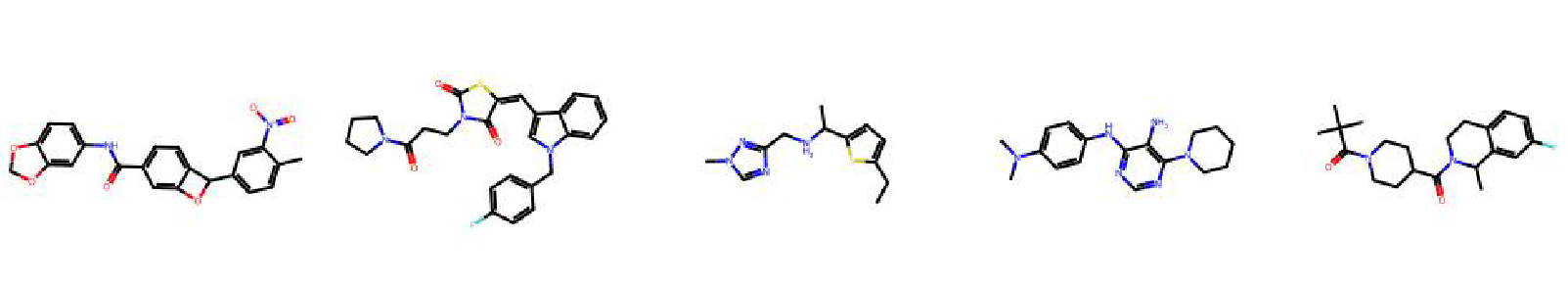
median1
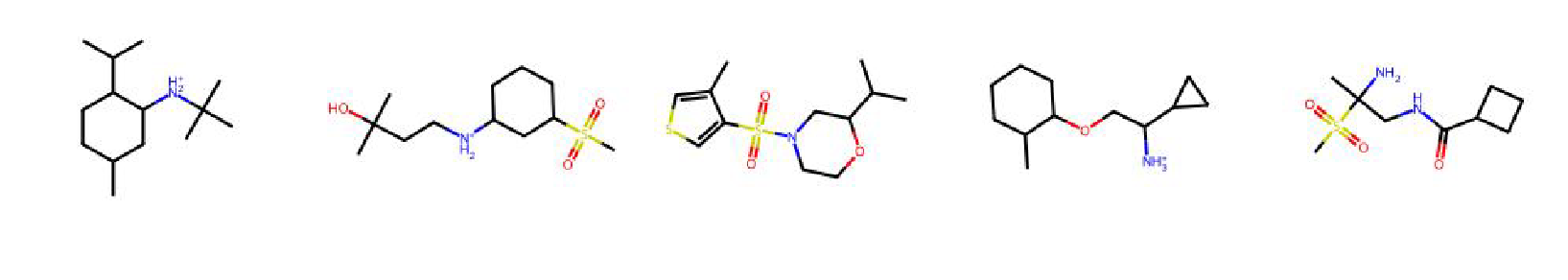
median2
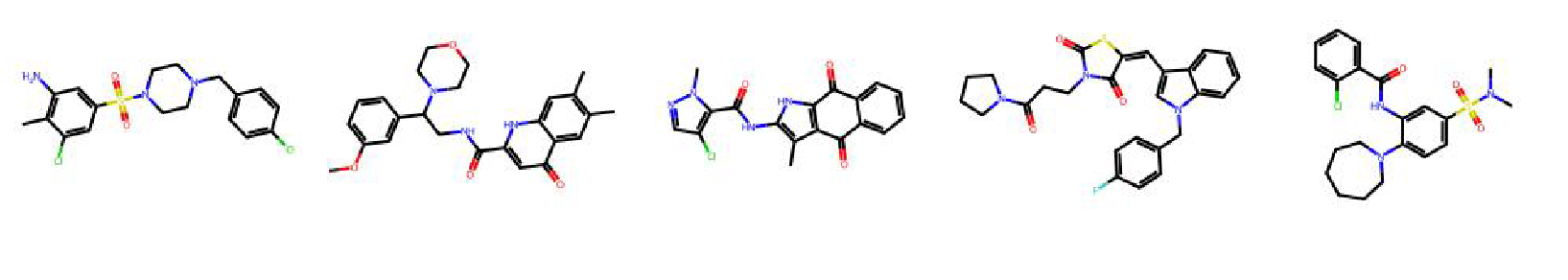
mestranol similarity
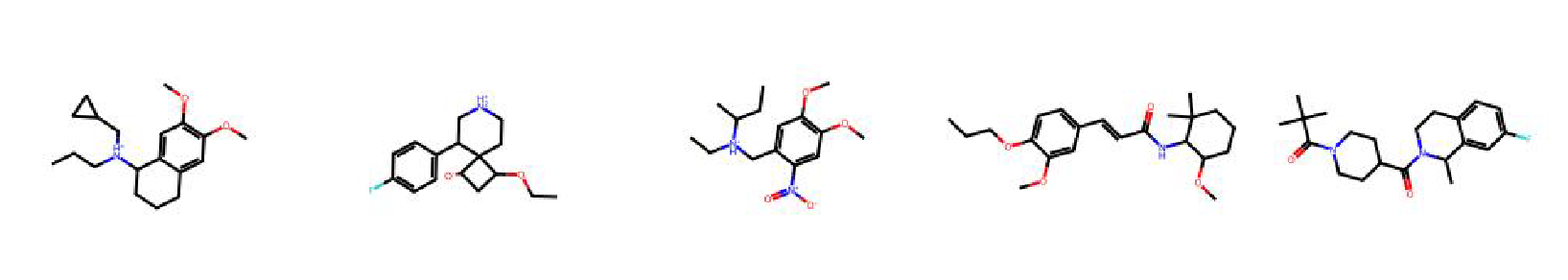
osimertinib mpo
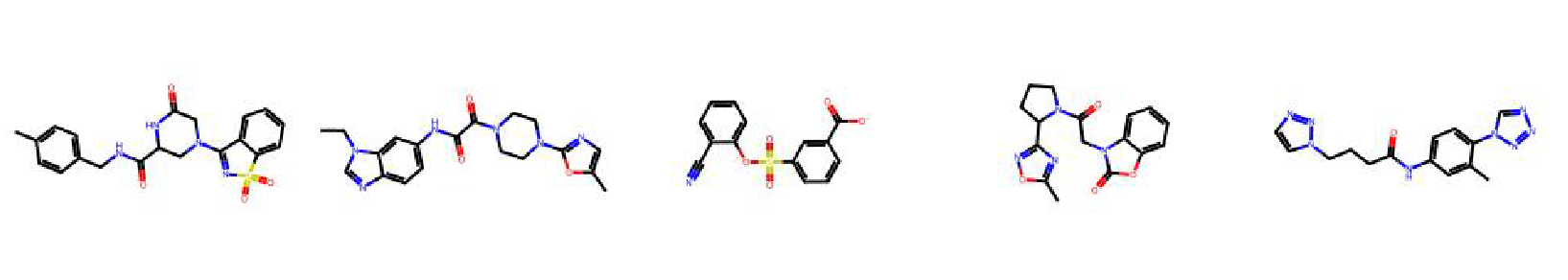
perindopril mpo
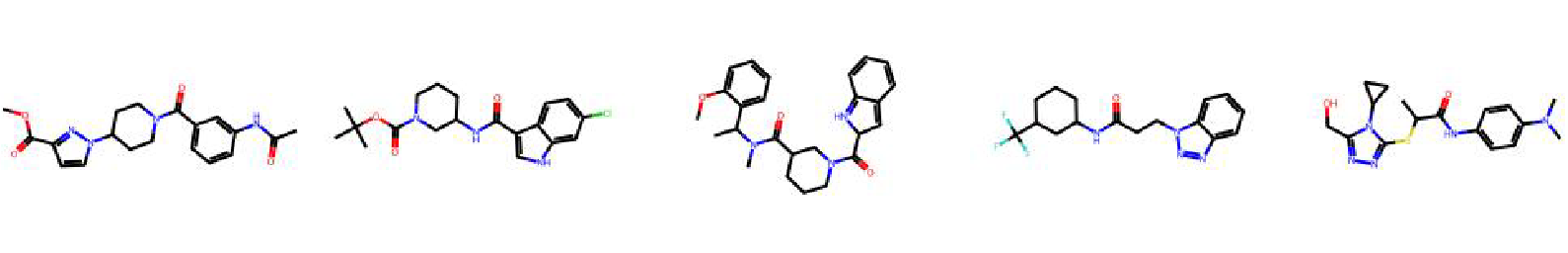
qed
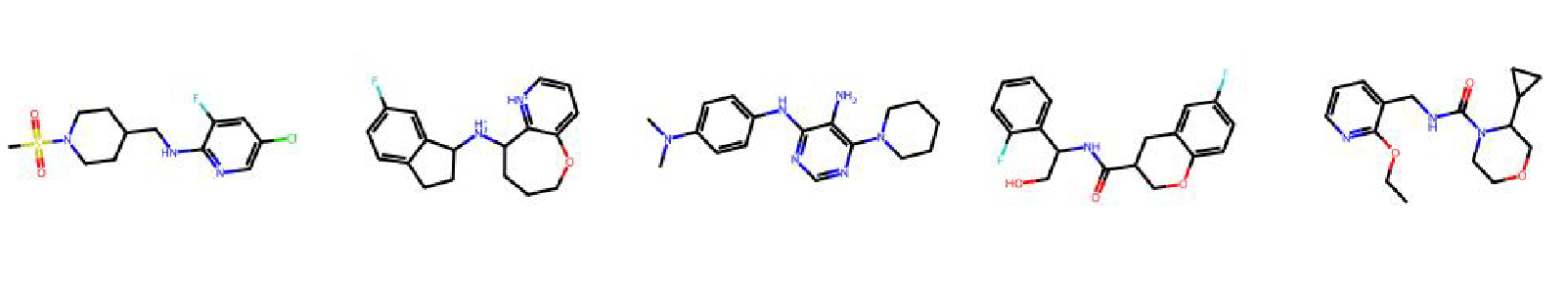
ranolazine mpo
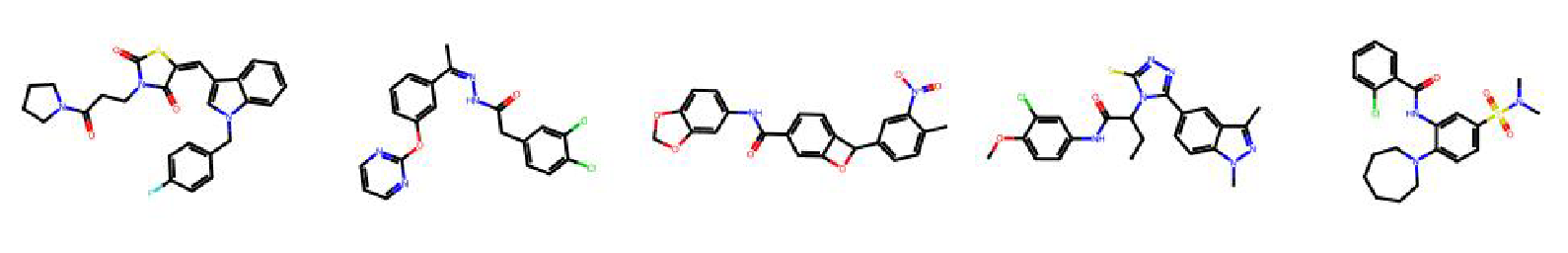
scaffold hop
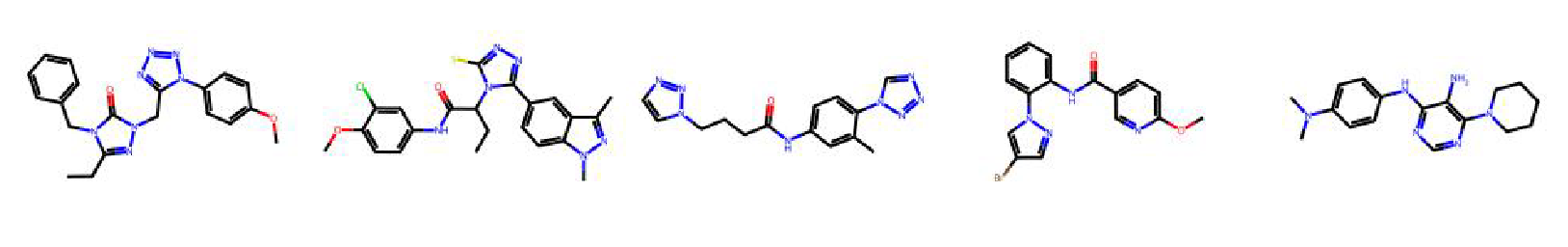
sitagliptin mpo
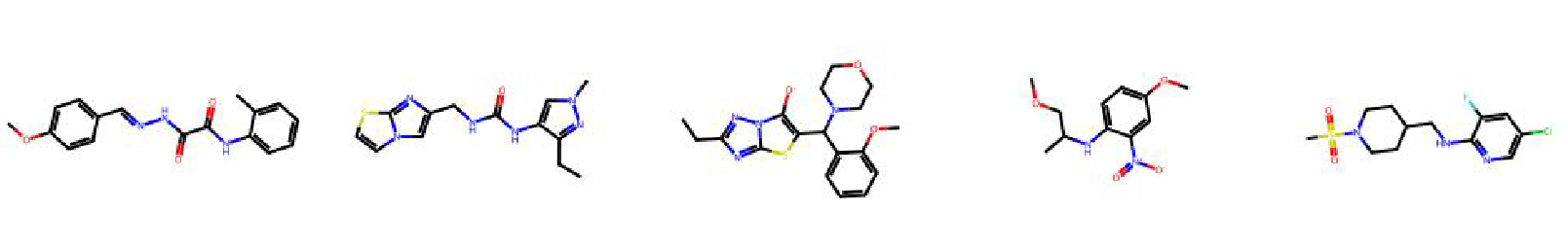
thiothixene rediscovery
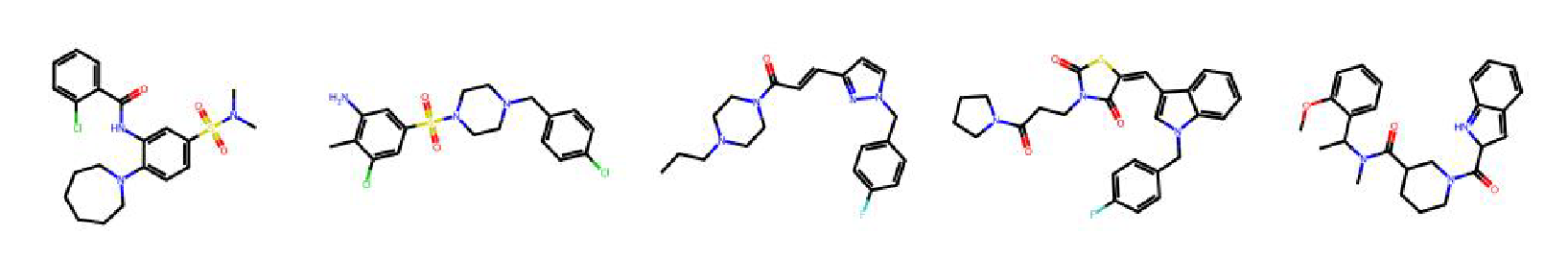
troglitazone rediscovery
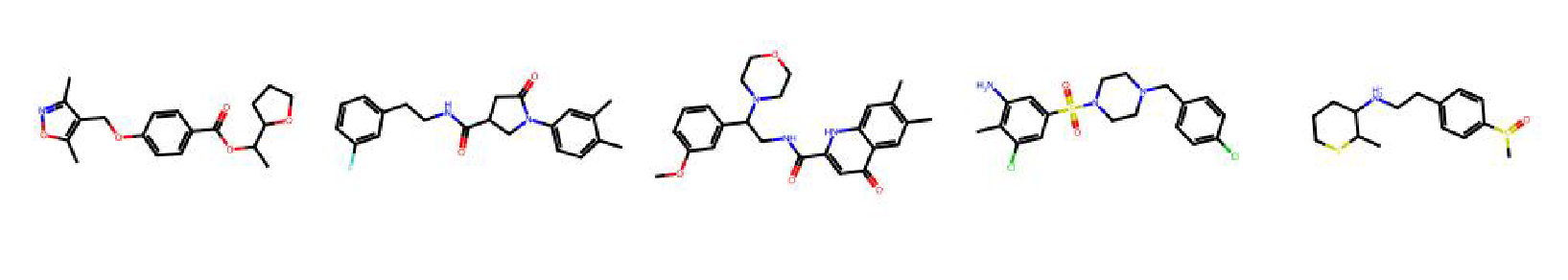
valsartan smarts
zaleplon mpo